Грибоподібний мікоз і синдром Сезарі
05.06.2020
Випадки з клінічної практики лікара-дерматопатолога Калмикової А.В.
Група Т-клітинних лімфом шкіри (ТКЛШ) – гетерогенна група неходжкінських лімфом, що представляють собою злоякісну трансформацію резидентних Т-лімфоцитів шкіри (табл. 1) [1]. Грибоподібий мікоз (mycosis fungoides – MF) та синдром Сезарі (СС) –найчастіші форми ТКЛШ, на їх частку припадає майже 65% усіх ТКЛШ. Зв’язок MF та СС довго залишався предметом дискусії. Раніше вважали, що СС є лейкемічною формою MF, але новітні дослідження довели, що це незалежні нозології. MF – це клональна проліферація резидентних ефекторних Т-лімфоцитів пам’яті шкіри, а СС є пухлинною трансформацією центральних Т-лімфоцитів пам’яті [2].

Грибоподібий мікоз і синдром Сезарі мають характерні, часто схожі, клінічні, морфологічні, імуногістохімічні та молекулярні характеристики. Однак якщо MF має індолентний клінічний перебіг з п’ятирічною виживаністю 88%, то СС належить до г рупи агресивних TКЛШ, і п’ятирічна виживаність при цьому захворюванні становить лише 24% [1]. Встановлення цих діагнозів зазвичай є непростим завданням і потребує покрокового вивчення стану пацієнта, ретельного клініко-морфологічного оцінювання та диференційної діагностики, а також проведення низки цитоморфологічних і молекулярних досліджень (табл. 2) [3-6]. Грибоподібний мікоз Грибоподібний мікоз є найчастішою формою ТКЛШ і становить майже 50% у структурі всіх ТКЛШ. Пухлина складається з малих/середніх Т-лімфоцитів, що характеризуються тропністю до епідермісу (епідермотропізмом). За визначенням це захворювання має стадійний перебіг (табл. 3) і характеризується еволюцією висипу – від плям до бляшок і пухлин [1, 3]. Як правило, це захворювання уражає осіб дорослого та похилого віку (медіана 55-60 років), але може виявлятися також у дітей та підлітків. Чоловіки хворіють дещо частіше за жінок (1,5-2:1). Патогенез захворювання невідомий.
рупи агресивних TКЛШ, і п’ятирічна виживаність при цьому захворюванні становить лише 24% [1]. Встановлення цих діагнозів зазвичай є непростим завданням і потребує покрокового вивчення стану пацієнта, ретельного клініко-морфологічного оцінювання та диференційної діагностики, а також проведення низки цитоморфологічних і молекулярних досліджень (табл. 2) [3-6]. Грибоподібний мікоз Грибоподібний мікоз є найчастішою формою ТКЛШ і становить майже 50% у структурі всіх ТКЛШ. Пухлина складається з малих/середніх Т-лімфоцитів, що характеризуються тропністю до епідермісу (епідермотропізмом). За визначенням це захворювання має стадійний перебіг (табл. 3) і характеризується еволюцією висипу – від плям до бляшок і пухлин [1, 3]. Як правило, це захворювання уражає осіб дорослого та похилого віку (медіана 55-60 років), але може виявлятися також у дітей та підлітків. Чоловіки хворіють дещо частіше за жінок (1,5-2:1). Патогенез захворювання невідомий.
Клінічні прояви Важливим клінічним проявом захворювання є прогресування висипу від плям до бляшок і пухлин. Перебіг захворювання досить індолентний, може тривати роками та навіть десятиріччями. До встановлення діагнозу пацієнти, як правило, мають історію неспецифічного екзематозного чи псоріазоформного дерматиту з неспецифічними гістологічними характеристиками. Медіана часу від перших клінічних проявів до встановлення діагнозу становить 4-6 років, але може варіювати від декількох місяців до 50 років [6, 7]. Початкові прояви MF характеризуються наявністю різних за розміром еритематозних плям із дрібнопластинчастим лущенням, можливий незначний свербіж. Ці ураження можуть мати легкий ступінь атрофії та зміни, властиві для пойкілодермії. Гіпопігментовані вогнища можуть спостерігатися в осіб з темною шкірою, це є частим симптомом ювенільного MF. Як правило, висип розташовується на сідницях, тулубі та кінцівках. З прогресуванням захворювання плями перетворюються на бляшки, що мають насиченіший червоно-коричневий колір, анулярну, поліцентричну або підковоподібну форми та пластинчасте лущення на поверхні. Якщо захворювання не має всіх клінічних складових і дебютує з пухлини, діагноз MF є сумнівним. При великоклітинній трансформації MF характерною є поява виразок на поверхні пухлин. Ризик ураження інших органів корелює зі стадією захворювання і є вищим у пацієнтів з еритродермією чи наявністю пухлин. Позашкірна дисемінація починається з регіонарних лімфатичних вузлів, що дренують найбільш уражені ділянки шкіри. Ураження кісткового мозку є нехарактерним для MF і відбувається нечасто. Морфологічні зміни Початкові прояви захворювання характеризуються наявністю в поверхневих відділах дерми щільного ліхеноїдного лімфоцитарного інфільтрату з ознаками епідермотропізму та формуванням в епідермісі невеликих скупчень клітин із типовими «церебриформними» ядрами (мікроабсцеси Потріє). Пухлинні лімфоцити, які містяться в епідермісі, трохи більші за клітини дермального інфільтрату та мають навколо себе світле гало. Уздовж дермо-епідермального з’єднання може спостерігатися формування ланцюжків пухлинних клітин. В епідермісі може визначатись акантоз, при цьому спонгіоз, як правило, не спостерігається. При прогресуванні захворювання інфільтрат може поширюватися на всі шари дерми з ураженням підшкірної жирової клітковини, епідермотропізм стає менш вираженим або навіть зникає. Великоклітинна трансформація MF характеризується появою великих атипових клітин у пухлинному інфільтраті, що експресують на своїй поверхні CD30 (близько 25% клітин пухлини). Цей феномен асоціюється з поганим перебігом захворювання. Імунофенотип Пухлинні клітини мають фенотип α/β Т-хелперів пам’яті (βF1+ TCRγ- CD3+ CD4+ CD5+ CD45RO+ CD8- TIA-).
У рідкісних випадках може спостерігатися фенотип цитотоксичних Т-лімфоцитів (βF1+ TCRγ- CD3+ CD4- CD5+ CD45RO+ CD8+ TIA+) або γ/δ Т-лімфоцитів (βF1- TCRγ+ CD3+ CD4- CD5+ CD45RO+ CD8+ TIA+), що не впливає на клінічний перебіг чи прогноз захворювання. У цих випадках кореляція з клінічною картиною є вкрай важливою для виключення агресивних цитотоксичних лімфом, таких як первинна агресивна епідермотропна CD8+ Т-клітинна лімфома чи γ/δ Т-клітинна лімфома шкіри. Втрата експресії пан-Т-клітинних маркерів (таких як CD7) є корисним діагностичним критерієм, але цей феномен нечасто спостерігається на початкових стадіях захворювання, тож його діагностичне значення не слід переоцінювати. Слід зазначити, що при деяких хронічних дерматитах може спостерігатися втрата пан-Т-клітинних маркерів. Диференційний діагноз 1. Хронічні дерматози, що можуть мати схожий клінічний перебіг (екзема, псоріаз, реакції на введення лікарських засобів тощо). 2. Хронічні дерматози, які можуть мати подібні морфологічні характеристики (лімфоматоїдний контактний дерматит, лімфоматоїдні реакції на введення лікарських засобів, актинічний ретикулоїд). 3. Інші епідермотропні ТКЛШ (агресивна епідермотропна CD8+ Т-клітинна лімфома, γ/δ Т-клітинна лімфома тощо).
 Клінічний випадок 1
Клінічний випадок 1
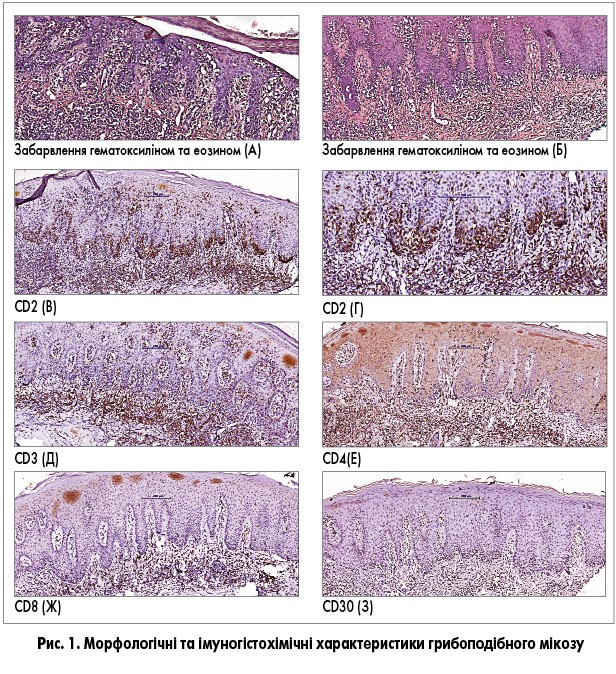
Пацієнт, 35 років, звернувся в лікарню зі скаргами на плями на шкірі, які спостерігалися протягом тривалого часу та суб’єктивно не турбували. Клінічний діагноз після проведення біопсії: Т-клітинна лімфома шкіри. Висип займав менше 10% площі шкіри. При біопсії в папілярному та ретикулярному шарах дерми виявлено щільний лімфоцитарний інфільтрат, що складався переважно з малих і середніх лімфоцитів зі значними ознаками епідермотропізму (рис. 1А-Г). Імунофенотип клітин інфільтрату – CD2+, CD3± (часткова втрата експресії), CD4± (часткова втрата експресії) та CD30-, CD8- (рис. 1В-З). Співвідношення CD4:CD8 становило приблизно 10:1 (рис. 1Е-Ж). Морфологічна картина, імунофенотип клітин пухлини, а також клінічні дані дали змогу діагностувати у пацієнта грибоподібний мікоз (pT1N0M0).
Синдром Сезарі
Синдром Сезарі становить 5% у структурі ТКЛШ, характеризується тріадою симптомів: еритродермією, генералізованою лімфаденопатією та наявністю пухлинних Т-лімфоцитів (клітин Сезарі) у шкірі, лімфатичних вузлах і периферичній крові. Критерії, рекомендовані для встановлення діагнозу СС: · наявність клітин Сезарі у периферичній крові; · виявлення клонального реаранжування ТКР у периферичній крові молекулярними чи цитогенетичними методами; · визначення імунофенотипічних порушень (проліферація CD4+ клітин призводить до порушення співвідношення CD4:CD8 більш ніж 10:1 – це аберантна експресія пан-Т-клітинних маркерів). У класифікації лімфом шкіри Всесвітньої організації охорони здоров’я та Європейської організації з вивчення та лікування онкологічних захворювань наявність Т-клітинного клону (бажано одного і того самого у шкірі та периферичній крові) в комбінації з зазначеними вище цитоморфологічними та імунофенотипічними критеріями визначаються як мінімальні критерії для діагностики СС і дозволяють виключити захворювання, що можуть мати подібний клінічний перебіг [1]. Патогенез синдрому Сезарі невідомий.
Клінічні прояви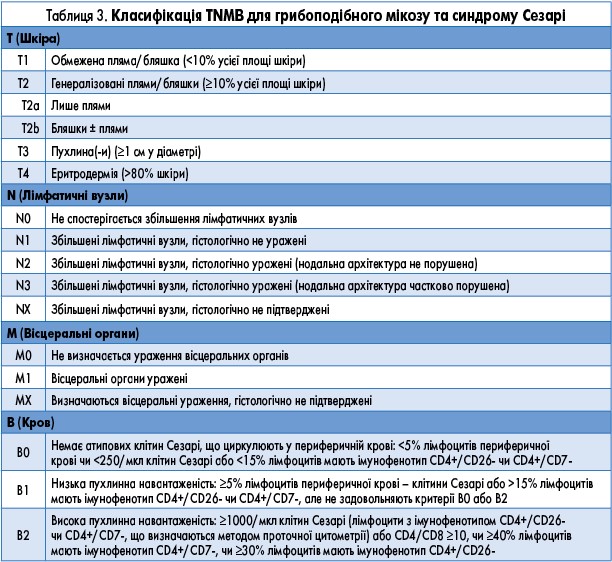
Синдром Сезарі характеризується еритродермією, що може бути асоційована зі значним лущенням, набряком, ліхеніфікацією та свербежем. Досить часто у пацієнтів спостерігаються лімфаденопатія, алопеція, оніходистрофія та пальмоплантарний гіперкератоз. СС належить до групи агресивних ТКЛШ з п’ятирічною виживаністю 24%. Більшість пацієнтів помирають від інфекційних ускладнень та імуносупресії.
Морфологічні зміни та імунофенотип Морфологічні зміни у шкірі при СС дуже схожі на зміни при MF, хоча при СС клітинний інфільтрат більш мономорфний, а епідермотропізм може бути відсутній. У третині випадків біоптати шкіри у пацієнтів з СС можуть мати неспецифічні гістологічні характеристики [8]. При ураженні лімфатичних вузлів спостерігається щільний дифузний інфільтрат, що складається з атипових клітин Сезарі зі «стиранням» нормальної архітектури лімфатичного вузла. При ураженні кісткового мозку, як правило, інфільтрація пухлинними клітинами не виражена, і клітини розташовуються переважно інтерстиційно. Імунофенотип пухлинних клітин: CD3+, CD4+, CD8- і (найчастіше) CD7- та CD26- [8].
Диференційний діагноз
Клінічний диференційний діагноз включає непухлинні еритродермії: псоріаз, атопічний дерматит, актинічний ретикулоїд, хворобу Девержі, ідіопатичну еритродермію, реакції на введення лікарських засобів. У цих випадках визначення клональності ТКР у периферичній крові є важливим діагностичним критерієм для СС. Враховуючи схожі клінічні та морфологічні характеристики СС та MF, необхідно також диференціювати ці два захворювання, оскільки лікування цих хвороб та прогноз при них значно відрізняються. Імунофенотип клітин інфільтрату – CD2+, CD5+, CD3+, CD4+, CD7+ та CD30-, CD8- (рис. 2Е-М). Співвідношення CD4:CD8 становило більше ніж 10:1 (рис. 2Ж, Л). Морфологічна картина, імунофенотип клітин пухлини, клінічні дані та результати лабораторних досліджень дозволили діагностувати у пацієнта синдром Сезарі (pT4NХсM0).
Останні новини


