Нотатки онколога: Синдром Лінча
22.11.2021
Близько 20% усіх випадків колоректального раку (КРР) носять спадковий характер.
Близько 20% усіх випадків колоректального раку (КРР) носять спадковий характер. Зважаючи на високу розповсюдженість спадкових форм КРР, обов’язковим етапом первинної діагностики даного виду раку є оцінка факторів ризику та виявлення даних обтяженого сімейного анамнезу щодо відповідних спадкових синдромів, серед яких найбільш часто зустрічається синдром Лінча.
Загальна інформація про синдром Лінча
Синдром Лінча (СЛ) – це найбільш розповсюджений спадковий синдром який обумовлює генетичну схильність до розвитку КРР, на частку якого припадає 2%-4% усіх випадків КРР. В загальній когорті пацієнтів з КРР даний синдром виявляється в 1 з 30 пацієнтів.
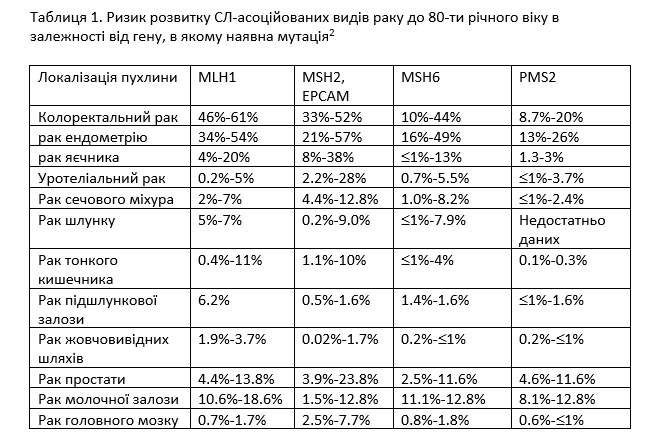
СЛ обумовлений наявністю або спадкових мутацій в одному з 4 генів сімейства MMR (MLH1, MSH2, MSH6, PMS2), або делецій в гені EPSAM які пригнічують транскрипцію гена MSH2. Найбільш розповсюдженими СЛ-асоційованими видами раку є КРР та рак ендометрію (РЕ). Асоційовані із СЛ КРР та РЕ характеризуються більш раннім віком постановки діагнозу (до 50 років), однак можуть виявлятися й в осіб старших вікових категорій (Таблиця 1).
Хірургічне лікування КРР у пацієнтів із синдромом Лінча
Пацієнти з СЛ-асоційованим КРР, в яких наявні мутації MLH1 або MSH2, знаходяться в групі підвищеного ризику розвитку метахронного КРР. З метою зменшення ризику розвитку метахронного КРР в даній когорті, при резекційних втручаннях з приводу первинного КРР, замість сегментальної резекції, можливе виконання колектомії в розширеному об’ємі; подальший план спостереження – колоноскопія прямої кишки 1 раз на рік. Рішення про об’єм оперативного втручання слід приймати на основі ризиків розвитку метахронного КРР, з врахуванням прогнозу та якості життя пацієнта. Після сегментальної резекції ризик розвитку метахронного КРР становить 16%, 41% та 62% через 10, 20 та 30 років, відповідно. За іншими даними, після сегментальної резекції даний показник становив 20% та 47% через 10 та 25 років, відповідно; після розширеної резекції – 4% та 9%, відповідно. Ризик розвитку метахронного КРР знижується на 31% на кожні 10 см видаленого кишечнику. Також розширений об’єм резекції може виступати в якості профілактики метахронного КРР у пацієнтів із СЛ, які не зможуть дотримуватися належного плану спостереження після завершення лікування з приводу первинного КРР (колоноскопія 1 раз на рік при мутаціях MLH1 або MSH2, 2 рази на рік – при мутаціях MSH6, PMS2, EPCAM).
Діагностика синдрому Лінча у пацієнтів з КРР
В тих випадках, коли в одного з близьких родичів пацієнта, факт наявності спадкової мутації в одному з 5 генів вже встановлено, для спростування або підтвердження діагнозу СЛ достатньо виконати дослідження на спадкові мутації в тому ж гені.
В осіб без інформації про відповідні спадкові мутації у родичів, на основі критеріїв сімейного анамнезу та в залежності від досліджуваного матеріалу, виділяють два основні шляхи діагностичного алгоритму СЛ:
1. В один етап - визначення спадкових мутацій за допомогою мультигенної панелі яка включає дослідження MLH1, MSH2, MSH6, PMS2 та EPCAM мутацій
2. В декілька етапів - рішення про проведення тесту на спадкові СЛ-асоційовані мутації приймається на основі результатів одного з наступних досліджень:
- імуногістохімічне дослідження (ІГХ) на виявлення білків 4 MMR генів (MLH1, MSH2, MSH6, PMS2)
- дослідження на статус мікросателітної нестабільності (MSI)
- NGS-панель яка включає щонайменше 4 MMR гени, гени EPCAM, BRAF, MSI та інші гени асоційовані із спадковими синдромами при КРР
У пацієнтів з КРР або з раком ендометрію (РЕ) перший шлях діагностичного алгоритму СЛ (мультигенна панель) може бути варіантом вибору при наявності одного з наступних критеріїв:
- Постановка діагнозу у віці до 50 років
- Синхронний/метахронний СЛ-асоційований рак у будь-якому віці
- 1 близький родич (першого/другого ступеня спорідненості) з СЛ-асоційованим раком в анамнезі у віці до 50 років
- Більше 2 близьких родичів із СЛ-асоційованим раком в анамнезі незалежно від віку постановки діагнозу
Для діагностики спадкових мутацій асоційованих з КРР, у пацієнтів з обтяженим сімейним анамнезом або іншими факторами які вказують на високий ризик генетичної схильності, перевага надається дослідженням на основі мультигенних панелей, оскільки при більш повному генетичному аналізі збільшується ймовірність виявити інші, в даному випадку не асоційовані із СЛ, спадкові мутації в даній когорті, що можуть мати клінічне значення як для пацієнтів, так і для їх родичів.
Другий шлях діагностичного алгоритму також може бути варіантом вибору у пацієнтів, які відповідають перерахованим вище критеріям. Окрім цього, діагностика СЛ в декілька етапів (перше дослідження або ІГХ, або MSI) виступає в якості скринінгового дослідження у пацієнтів з КРР або РЕ (Схема 1). 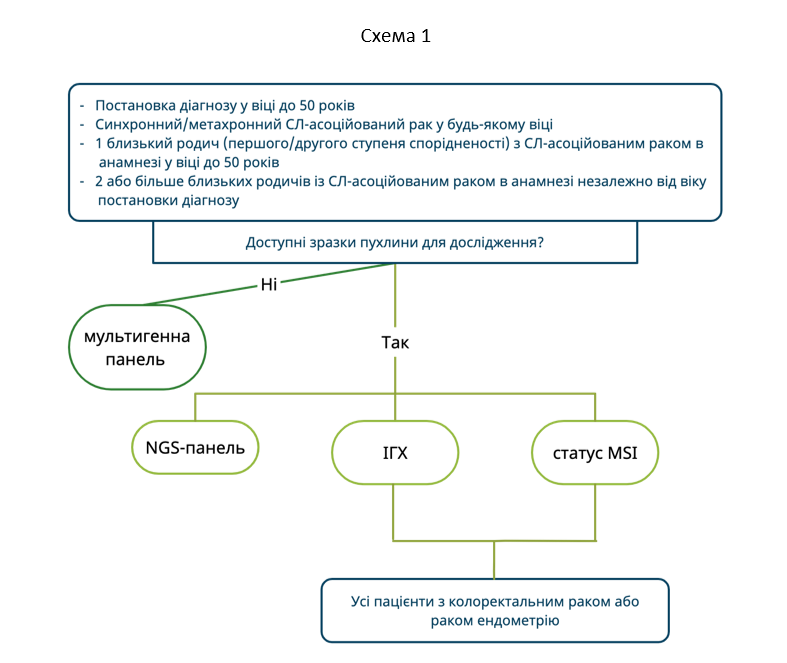
Скринінг СЛ є «універсальним», тобто, ІГХ або визначення MSI слід проводити усім пацієнтам з КРР/РЕ, оскільки чутливість критеріїв для виявлення пацієнтів підвищеного ризику становить 82%. Скринінг СЛ показаний усім пацієнтам з КРР/РЕ незалежно від віку постановки діагнозу. Попри те, що маніфестація СЛ-асоційованих видів раку, в більшості випадків, відбувається до 50 років, КРР/РЕ виявлені в більш старшому віці також можуть бути пов’язані із СЛ. При проведенні скринінгових досліджень лише в когорті пацієнтів з КРР до 50 років, СЛ не буде діагностовано більше, ніж в половині випадків. Альтернативним методом скринінгу у пацієнтів з КРР старше 70 років є проведення відповідних досліджень лише тим пацієнтам які відповідають вищевказаним критеріям.
По результатам одного зі скринінгових досліджень другого діагностичного алгоритму, про можливу наявність спадкових мутацій в MMR генах свідчить одне з наступних:
• ІГХ: відсутня експресія одного з 4 білків (свідчить про дефект в одному з MMR генів – dMMR)
• Статус MSI: високий ступінь мікросателітної нестабільності (MSI-H)
Кожне дослідження (ІГХ або MSI) може бути використано в якості першого етапу скринінгу СЛ і наразі немає загальноприйнятного консенсусу щодо того, якому з них слід надавати перевагу. MSI-H є критерієм для призначення імунотерапії пацієнтам з метастатичним КРР та може виступати в якості фактора ризику при призначенні хіміотерапії на ІІ стадії захворювання. В той же час, результати ІГХ допомагають більш точно визначити когорту пацієнтів яким показано проведення наступних досліджень на спадкові СЛ-асоційовані мутації.
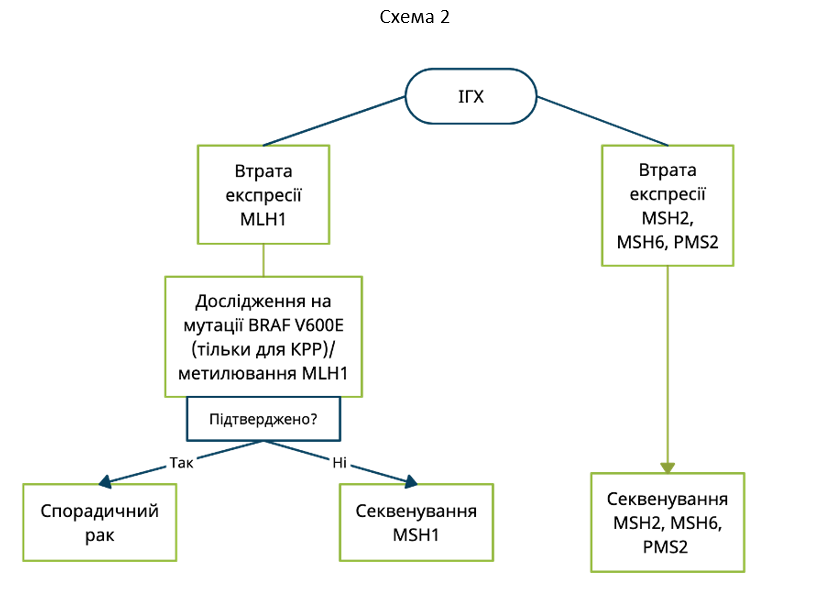
Втрата експресії MLH1 та/або PMS2 на ІГХ може вказувати на метилюванням гену MLH1 (мMLH1). мMLH1 зустрічається в 10-15% випадків спорадичних форм КРР. Для спростування або підтвердження мMLH1 можливим є проведення або дослідження пухлини на BRAF мутації (тільки для КРР), або проведення тестування на мMLH1. Наявність BRAF мутацій та/або мMLH1 виключає наявність спадкових СЛ-асоційованих мутацій – в таких випадках немає необхідності в проведенні додаткових генетичних досліджень (Схема 2).
Дослідження пухлини за допомогою NGS-панелі дозволяє отримати найбільш повноцінну інформацію щодо усіх можливих показників, які можуть вказувати на наявність спадкових мутацій. Однак, в якості скринінгу, дане дослідження не є економічно-вигідним. NGS-панель може бути варіантом вибору у пацієнтів з КРР які відповідають вищевказаним критеріям діагностики СЛ.
Останнім етапом другого діагностичного алгоритму СЛ є дослідження на спадкові СЛ-асоційовані мутації, що може здійснюватися або за допомогою секвенування вибіркових генів (MLH1, NSH2, MSH6, PMS2, EPCAM), або за допомогою мультигенної панелі (у пацієнтів з обтяженим сімейним анамнезом).
Автор: Марія Білич, лікар-онколог, член Української спілки клінічної онкології
1. Al B. Benson, Venook AP, Al-Hawary MM, Mwanzi SA, Al EN et. Colon Cancer Colon Cancer. NCCN Harmon Guidel Sub-Saharan Africa - Colon Cancer. 2018;Version 2.(Colon Cancer):1-5.
2. Burke CA, Dallas S. Genetic / Familial High-Risk Assessment : Colorectal. Published online 2021.
3. Mehta SR et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). New England Journal. N Engl J Med. 2015;687-696. doi: 10.1056/NEJMoa043146
4. Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69(3):411-444. doi:10.1136/gutjnl-2019-319915.
5. Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers – the advantage of more extensive colon surgery 2013;60(7). doi:10.1136/gut.2010.228056.
6. Renkonen-Sinisalo L, Seppälä TT, Järvinen HJ, Mecklin JP. Subtotal Colectomy for Colon Cancer Reduces the Need for Subsequent Surgery in Lynch Syndrome. Dis Colon Rectum. 2017;60(8):792-799. doi:10.1097/DCR.0000000000000802
7. Gudgeon JM, Belnap TW, Williams JL, Williams MS. Impact of age cutoffs on a lynch syndrome screening program. J Oncol Pract. 2013;9(4):175-179. doi:10.1200/JOP.2012.000573
Останні новини


