Клинические и морфологические особенности первичных CD30+ лимфопролиферативных заболеваний кожи
05.06.2020
Случаи из клинической практики доктора-дерматопатолога Калмыковой А. В.
Т-клеточные лимфомы кожи (ТКЛК) — это широкая группа заболеваний, включающая различные нозологии, характеризующиеся специфическими клиническими проявлениями, морфологическими изменениями и прогнозом.
Лимфоматоидный папулез (ЛП) и первичная анапластическая крупноклеточная лимфома кожи (ПК-АКЛ) являются составляющими одного патологического процесса и имеют сходные морфологические и иммуногистохимические характеристики, однако разную клиническую картину [1].

Наличие пограничных состояний, когда у пациента наблюдаются проявления обоих заболеваний, подтверждает эту теорию. Поэтому в новой классификации лимфом кожи [2] ЛП и ПК-АКЛ включены в группу первичных CD30+ лимфопролиферативных
заболеваний, характеризующихся экспрессией на поверхности клеток антигена CD30 и, несмотря на агрессивные морфологические признаки, имеют благоприятный прогноз. CD30 также экспрессируется при ряде других ТКЛК, поэтому диагностика таких состояний требует полного иммунофенотипирования клеток инфильтрата и тщательной клинико-морфологической корреляции.
 Клинический случай 1
Клинический случай 1
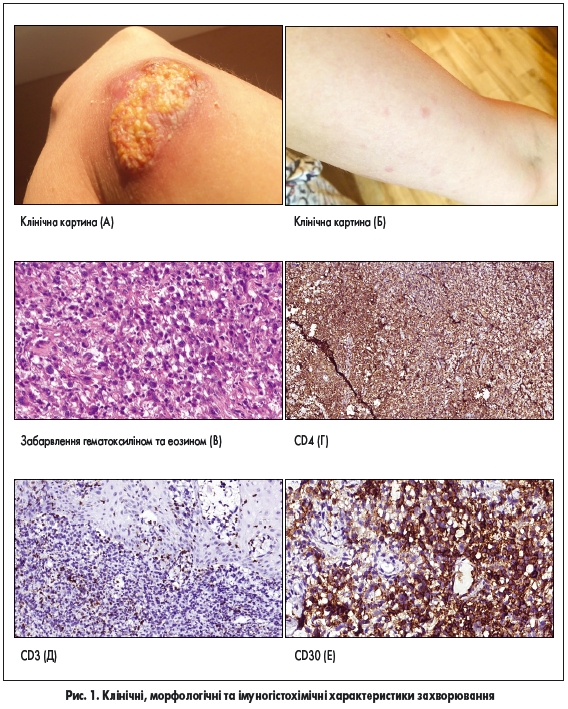
Пациентка, 32 года, обратилась в больницу с жалобами на опухоль с язвой (рис. 1А), возникшую несколько месяцев назад. Пациентка также отмечала появление папул и узлов (рис. 1Б), которые иногда сопровождались изъязвлением, а впоследствии самостоятельно заживали с образованием нормотрофических рубцов. Общее состояние пациентки на момент осмотра удовлетворительное. Больной проведена биопсия, по результатам которой выявлены следующие изменения (рис. 1В) в дерме наблюдался отек и диффузный лимфоцитарный инфильтрат из малых и средних лимфоцитов со значительной примесью больших клеток, которые имели гиперхромные большие ядра, определялось большое количество фигур митозов. Данные иммуногистохимического исследования: клетки опухоли CD4, CD30-позитивные, часть клеток — CD3-положительные и ALK-отрицательные (последние не представлены на рисунке) (рис. 1Г‐Е).
После того, как на основании клинико-лабораторных данных была исключена системная АКЛ, по морфологическим изменениям и иммунофенотипом клеток опухоли диагностирована ПК-АКЛ, однако, учитывая клиническое течение и наличие сыпи, которая самостоятельно заживала и появлялся снова, этот случай заболевания можно отнести к пограничным состояниям, когда у пациента имеются проявления как ПК-АКЛ, так и ЛП. Клинический случай 2
Клинический случай 2
Пациентка, 52 года, имела жалобы на папуло-некротическую сыпь на туловище, которая субъективно не беспокоила (рис. 2А‐Б), отмечала появление новых элементов сыпи с одновременным регрессированием старых. Клинический диагноз — pityriasis lichenoides et varioloformis acuta. Морфологические признаки заболевания — сливной пара- и гиперкератоз, определялись лейкоциты в паракератотических массах. В базальных отделах эпидермиса наблюдался интерфейс-дерматит с наличием цитоидных телец и сателлитных некрозов кератиноцитов (рис. 2в). При иммуногистохимическом исследовании среди клеток инфильтрата обнаружены CD30+ клетки (рис. 2Г). На основании наличия характерной клинической картины и морфологических изменений установлен диагноз лимфоматоидного папулеза (тип В).
Клинический случай 3
Пациентка, 72 года, обратилась в больницу с жалобами на пятна на коже, которые наблюдались в течение длительного периода и субъективно не беспокоили до последнего времени, однако несколько месяцев назад на месте пятен появились сначала бляшки, а впоследствии — язвы (рис. 3А‐Б). При биопсии в дерме наблюдался плотный лимфоцитарный инфильтрат преимущественно из малых и средних лимфоцитов со значительной примесью крупных гиперхромных клеток, которые размещались как в поверхностных, так и в глубоких отделах дермы с распространением в подкожную жировую клетчатку (рис. 3В‐Г). Определен выраженный эпидермотропизм клеток
инфильтрата с формированием в эпидермисе мелких скоплений (микроабсцессов Потрие) (рис. 3В). Иммунофенотип клеток инфильтрата — CD3+, CD4+ и CD30+, CD7-, CD8-, ALK-. Примерно 80 % клеток опухоли Ki-67-положительные (маркер пролиферации; рис. 3Д‐М). На основании имеющейся морфологической картины и определения иммунофенотипа клеток опухоли, а также клинических данных диагностирована крупноклеточная трансформация грибовидного микоза.
Обсуждение
ТКЛК — широкая группа заболеваний, включающая различные нозологии, характеризующиеся специфическими клиническими проявлениями, морфологическими изменениями и прогнозом [2] (табл. 1).
Наиболее распространенными в группе ТКЛК являются грибовидный микоз и синдром Сезари, которые составляют примерно 65 % всех случаев. CD30+лимфопролиферативные заболевания, такие как ЛП и ПК-АКЛ — около 25 %. Частота других лимфом, которые встречаются значительно реже, составляет лишь 10 % случаев (табл. 2).
В зависимости от клинического течения ТКЛК разделяют на агрессивные и индолентные (табл. 3).
Несмотря на то что грибовидный микоз относится к группе индолентных лимфом, при крупноклеточной трансформации прогноз этого заболевания значительно ухудшается [4]..
Первичные CD30 + лимфопролиферативные заболевания кожи
Первинні CD30+ лімфопроліферативні захворювання шкіри складають другу за поширеністю групу нозологій серед ТКЛШ [2] (табл. 2). До них належать ПШ-АВЛ, ЛП та пограничні стани.ЛП та ПШ-АВЛ можуть мати спільні клінічні, морфологічні та імуногістохімічні ознаки. На даному етапі вивчення вважається, що ці захворювання складають спектр одного патологічного процесу [5]. Для встановлення морфологічного діагнозу пацієнтам із підозрою на первинні CD30+ лімфопроліферативні захворювання шкіри необхідно проводити біопсію з подальшим імуногістохімічним дослідженням та за необхідності молекулярно-генетичне дослідження для встановлення клональності клітин інфільтрату.
Лимфоматоидный папулез
Впервые это заболевание описал W. L. Macaulay в 1968 году [6]. В структуре заболеваемости ТКЛК доля ЛП составляет 15 %. ТКЛК поражает лиц всех возрастных групп, средний возраст пациентов составляет 35–45 лет. У мужчин заболевание регистрируют чаще, чем у женщин (1,5:1). ЛП — хроническое доброкачественное самолимитирующееся заболевание, характеризующееся папулезной, папуло-узловой или папуло-некротической сыпью и имеет рецидивирующий характер. Сыпь локализована, как правило, диссеминировано на конечностях и туловище пациента, однако может иметь и сгруппированный характер, ограничиваясь одной локализацией. Цвет элементов сыпи варьирует от красного до фиолетового, размер — от нескольких миллиметров до 2 см. Поскольку сыпь имеет рецидивирующий характер, со временем у пациента начинает наблюдаться полиморфизм элементов. Примерно в 50 % случаев заболевания сыпь субъективно не беспокоит, но иногда присутствует зуд и ощущение жжения. Обычно сыпь исчезает в течение 1–4 мес, но может наблюдаться в течение длительного времени — несколько месяцев и даже лет, самый долгий зафиксированный период его наличия составлял около 40 лет [7].
Патогенез ЛП пока не выяснен. Несколько авторов выдвинули предположение о вирусной природе этого заболевания, однако по результатам ряда исследований не установлена роль Т-лимфотропного вируса человека типа 1 (HTLV‐1), вируса Эпштейна — Барр (EBV) и других вирусов герпесвирусной группы в патогенезе ЛП. Механизм спонтанной регрессии сыпи, который наблюдается у части пациентов, до сих пор не установлен. Наиболее вероятно, причиной этого феномена является взаимодействие CD30 с лигандом CD30, что приводит к апоптозу опухолевых Т-клеток [8]. Выделяют 5 основных морфологических типов ЛП — типы A–E. Недавно были описан: 6‐й — тип F и редкие типы, такие как γ/δ-тип ЛП с реаранжировкой генов 6р25 [10]. При иммуногистохимическом исследовании в большинстве случаев определяются клетки инфильтрата CD45RO+ и CD4+. Все формы заболевания характеризуются наличием CD30+ клеток в инфильтрате, за исключением типа В, который требует тщательной дифференциальной диагностики с грибовидным микозом. В этом случае важным является проведение клинико-морфологической корреляции. Типы D, E ЛП у детей могут быть CD4-, CD8+. Важно дифференцировать ЛП, который является CD45RO- положительным, с эпидермотропной CD8+ ТКЛК, которая обычно CD45RО- [2]. Кроме этого, при проведении дифференциальной диагностики следует учитывать возможность наличия состояний, перечисленных в таблице 4. Следует отметить, что характер клинического течения не зависит от морфологического подтипа заболевания. Его определение имеет важное значение для проведения дифференциальной диагностики и оценки прогноза, поскольку подтипы В и С ассоциированы с развитием вторичных гематологических опухолей [11].
Первичная анапластическая крупноклеточная лимфома кожи
ПК-АКЛ характеризуется наличием солитарной опухоли размером >2 см в диаметре, которая быстро растет и сопровождается изъязвлением. Иногда случаются круноочаговые формы заболевания. Несмотря на агрессивное клиническое течение ПК-АКЛ, пациенты имеют хорошее самочувствие, у них не наблюдаются симптомы системного поражения (температура, слабость, повышенная потливость, снижение массы тела и др.). Наличие этих симптомов имеет большое значение, так как крайне важным в диагностике ПК-АКЛ является исключение возможного вторичного поражения кожи вследствие системной АКЛ. Около 20 % пациентов с системной АКЛ имеют очаги вторичного поражения кожи [12].
При морфологическом исследовании заболевание характеризуется плотным лимфоцитарным инфильтратом с анапластической морфологией: клетки имеют обильную
эозинофильную цитоплазму и большие, плеоморфные, гиперхромные ядра, некоторые имеют форму подковы и большое эозинофильное ядрышко. Для установления диагноза ПК-АКЛ менее 75 % клеток опухоли должны быть CD30-положительными. При кожных формах заболевания киназа анапластических лимфом (ALK) обычно негативная, однако это не должно быть решающим признаком при дифференцировании с системной АКЛ, поскольку около 50 % этих опухолей является ALK- [13]. Кожный лимфоцитарный антиген (CLA) при ПК-АКЛ, как правило, положительный, зато эпителиальный мембранный антиген (EMA) — отрицательный [14]. Проводить дифференциальную диагностику необходимо с системной АКЛ, ЛП, крупноклеточной трансформацией грибовидного микоза и другими лимфомами, клетки которых могут экспрессировать на своей поверхности CD30 антиген (в том числе с вторичным поражением кожи при лимфоме Ходжкина), а также с реактивными воспалительными реакциями.
Пограничные состояния
К пограничным состояниям относятся случаи заболевания, если дифференцировать ЛП и ПК-АКЛ невозможно. По результатам тщательного и длительного наблюдения пациента можно более точно определить, какое из этих заболеваний имеет место.
 Прогноз заболевания
Прогноз заболевания
Первичные CD30+ лимфопролиферативные заболевания кожи имеют благоприятный прогноз. Десятилетняя выживаемость составляет 85–100 % [14]. При ведении таких пациентов следует учитывать, что ЛП может быть ассоциирован с развитием вторичных гематологических опухолей. Чаще всего наблюдается грибовидный микоз и АКЛ [11]. В зависимости от диагностированной формы заболевания клиническая тактика при лечении ТКЛК и прогноз значительно отличается по сравнению с нодальной формой. Например, в отличие от последней, ПК-АКЛ не требует агрессивного лечения, поэтому установление точного диагноза является залогом выбора правильной тактики ведения пациента.
Общие сведения об антигене CD30
Антиген CD30 (кластер дифференцировки 30) — трансмембранный гликопротеин 1 типа,который относится к суперсемейству рецепторов фактора некроза опухоли 8 (TNFRSF8). Впервые этот рецептор был описан в клетках лимфомы Ходжкина, впоследствии аналогичный антиген обнаружен в ряде неходжкинских лимфом, в том числе ПК-АКЛ. CD30 — мембранный цитокиновый рецептор, который появляется на поверхности митогенактивированных Т- и некоторых В-лимфоцитов. Экспрессия CD30 требует активации сигнального пути CD28 или рецептора интерлейкина‐4 [15, 16]. CD30 взаимодействует с лигандом CD30 (CD30L, CD153, TNFSF8) — мембранным
гликопротеином 2 типа, также относится к суперсемейству TNF и экспрессируется на активированных Т-лимфоцитах, преимущественно на поверхности CD4+ клеток — Т- хелперах 1 и 2 типа, а также на поверхности B-лимфоцитов и других иммунокомпетентных клеток, в том числе антигенпрезентующих клеток [17]. Активация сигнального пути CD30 приводит к активации ядерного фактора NF‐kB как путем активации TNF-рецептор-ассоциированного фактора 2 (TRAF), так и через независимый от этого фактора путь, который может подавлять активность эффекторных клеток, стимулировать апоптоз или, наоборот, приводить к выживаемости клеток в зависимости от типа привлеченных к активации клеток и активированных сигнальных путей [18, 19]. Экспрессию CD30 на поверхности лимфоцитов могут вызывать целый ряд факторов, в
том числе реактивные состояния, воспалительные и инфекционные заболевания, опухолевые процессы, поэтому для установления диагноза необходимы тщательная клинико-морфологическая корреляция, проведение морфологического и иммуногистохимического исследования биоптатов кожи, а в некоторых случаях — молекулярно-генетического исследования по определению клональности Т-клеток инфильтрата [20] (табл. 4).
Выводы
ТКЛК — широкая группа заболеваний, включающая различные нозологии, которые характеризуются специфическими клиническими проявлениями, морфологическими изменениями и прогнозом. ЛП и ПК-АКЛ составляют спектр одного патологического
процесса и имеют сходные морфологические и иммуногистохимические характеристики, однако разную клиническую картину. CD30 — важный маркер для диагностики и классификации ЛК, однако интерпретацию положительной реакции следует осуществлять в комплексе с точными иммунофенотипичными характеристиками опухоли и исчерпывающими клиническими данными.
Несмотря на то что большинство ТКЛК имеют индолентное клиническое течение, своевременное установление точного диагноза должно быть правилом, а не исключением. Без полных клинических данных проведение дифференциальной диагностики может быть затруднено или даже невозможно, поэтому клинико-морфологическая корреляция является критически важной в ведении пациентов с подозрением на ТКЛК.
В течение последних нескольких лет наблюдается значительный прогресс с точки зрения понимания клинического течения, патологических характеристик, биологического потенциала и этиологических факторов группы заболеваний, к которой принадлежит ТКЛК. Несмотря на это, до сих пор много невыясненных вопросов, в частности, активно дискутируется классификация, этиологические факторы и прогностические признаки этих заболеваний.
Последние новости


